Así se vigilan los fármacos una vez aprobados
Las condiciones reales en las que se usa un fármaco son distintas de las condiciones controladas en las que se ha llevado a cabo el ensayo clínico.


“¿Alguno de sus lectores ha observado anomalías similares en bebés nacidos de madres que tomaban este fármaco durante el embarazo?”
Con esta frase W.G. McBride, médico australiano, terminaba hace 60 años una carta en la que expresaba una preocupación y trataba de confirmarla preguntando a otros colegas (Mcbride, 1961). El fármaco al que se refería era la talidomida y las anomalías, la focomelia. Otros sospecharon lo mismo. Y también lo comunicaron. Finalmente, la tragedia se confirmó.
Este desastre farmacológico sentó las bases de la farmacovigilancia actual y, aunque existen otros métodos, la comunicación de sospechas de reacciones adversas a fármacos sigue siendo la mejor manera de identificar problemas de salud causados por medicamentos, las llamadas reacciones adversas.
El incidente con la talidomida también hizo que se endurecieran los requisitos necesarios para autorizar los medicamentos. A día de hoy, deben ser evaluados primero en laboratorio y en animales (ensayos preclínicos) y después en humanos (ensayos clínicos), antes de ser autorizados para su utilización.
Los criterios para autorizarlos son muy rigurosos y persiguen un fin: que el fármaco tenga un equilibrio entre el beneficio que va a producir y sus posibles riesgos. Favorable, por supuesto, al beneficio. Solo cuando esto se demuestra se puede autorizar el fármaco.
Por desgracia, aunque son lo mejor que tenemos, los ensayos clínicos no son perfectos. Ciertas limitaciones impiden que podamos identificar algunos problemas, como por ejemplo los que son poco frecuentes. Además, por motivos éticos, algunos pacientes no se suelen incluir en los ensayos clínicos. Es el caso de los niños, los ancianos o las mujeres embarazadas.
En definitiva, las condiciones reales en las que se va a usar un fármaco son distintas de las condiciones controladas en las que se ha llevado a cabo el ensayo clínico.
El fármaco está aprobado, ha demostrado ser eficaz y seguro en la indicación para la que se va a usar, y comienza a utilizarse en las condiciones reales. ¿Cómo se llevan a cabo las tareas de farmacovigilancia a partir de ese momento?
Cuando un profesional sanitario sospecha que un fármaco puede estar causando un problema de salud a un paciente, debe notificarlo a su Centro Autonómico de Farmacovigilancia. Esta notificación, que de forma tradicional se ha llevado mediante un formulario en papel (la Tarjeta Amarilla), puede hacerse desde el año 2017 a través de un formulario online en www.notificaram.es. Y por sí sola no basta para tomar ninguna decisión.
Es más, desde ese mismo año (2017) cualquier ciudadano puede notificar sus sospechas de reacciones adversas a medicamentos. Eso sí, los profesionales sanitarios están obligados pero los ciudadanos no.
Esto es lo que estamos viendo con las vacunas frente a la covid-19. Concretamente, con respecto a las vacunas y con el fin de recoger más información, se promueve la notificación de cualquier problema ocurrido tras la vacunación (asociación temporal). Pero para cada caso individual es casi imposible saber si el medicamento o vacuna ha sido el causante. No hay pruebas específicas para esto, exceptuando para el caso concreto de las alergias. Hace falta tiempo, más notificaciones, y estudios adecuados, para conocer con qué probabilidad el fármaco podría causar la reacción.
Solo ante notificaciones de varios profesionales sanitarios con la misma reacción adversa se genera una alerta a partir de la cual se comienza a investigar.
La farmacovigilancia es una de esas actividades profesionales que suelen pasar inadvertidas. En el Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano, con un Centro de Farmacovigilancia en cada Comunidad Autónoma y coordinados desde la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS), trabajan decenas de técnicos de farmacovigilancia. Su trabajo consiste en identificar estas posibles alertas: reacciones o problemas que no se han visto durante los estudios previos a la autorización.
Todos los casos notificados en España pasan a formar parte de la base de datos del SEFV-H: FEDRA (Farmacovigilancia Española Datos de Reacciones Adversas) y también de la base de datos europea: Eudravigilance. Un comité europeo, el Pharmacovigilance Risk Assessment Committee_ (PRAC) se encargará de evaluar las alertas identificadas.
La clave está en ese balance entre el beneficio y el riesgo. ¿Aceptamos los mismos riesgos para un fármaco contra la tos que para un fármaco con el que vamos a tratar un cáncer? Es obvio que no.
Si después de autorizarse un medicamento se identifica un riesgo nuevo que no es aceptable en relación al beneficio que produce, ese fármaco se retirará del mercado. La mayor parte de las veces el riesgo no implica la retirada, pero sí alguna acción informativa como notas de seguridad, modificaciones del prospecto y la ficha técnica del fármaco, restricciones de uso, etc.
Lo que no se discute es que todos los fármacos pueden producir reacciones adversas. Está en su propia naturaleza y es algo que, aunque tratemos de minimizar, al menos de momento, es inevitable.
¿Por qué? Porque los fármacos son sustancias que modifican las funciones del organismo, y no se ha conseguido una selectividad tal que solo actúen dónde y cuándo queremos. En consecuencia, de forma inevitable, otras funciones u otros órganos distintos a los deseados se verán también afectados, dando lugar a efectos no deseados y, en ocasiones, nocivos.
Es más, hasta el 5,3 % de los ingresos hospitalarios pueden estar relacionados con reacciones adversas a medicamentos, el 10 % en el caso de ancianos, siendo los analgésicos y antiinflamatorios la medicación más relacionada con estos últimos casos.
Esos medicamentos que nos curan, mejoran nuestra calidad de vida y la prolongan, puntualmente también pueden dar problemas. La farmacovigilancia es esencial para poder tener medicamentos seguros y utilizarlos de la mejor forma posible.





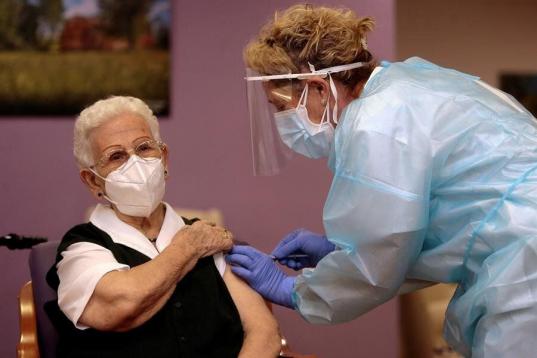

Araceli y Mónica, primeras personas en ser vacunadas contra el coronavirus en España.

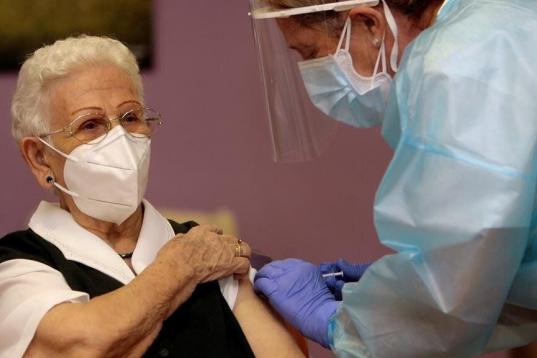
Araceli y Mónica, primeras personas en ser vacunadas contra el coronavirus en España.

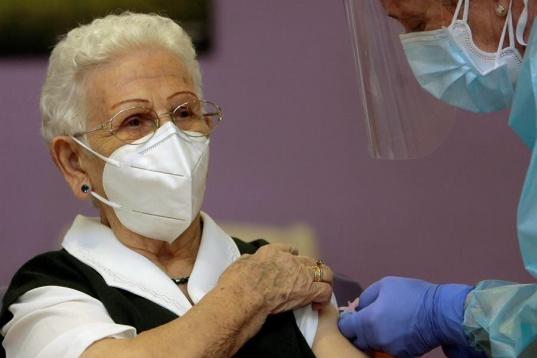
Araceli y Mónica, primeras personas en ser vacunadas contra el coronavirus en España.

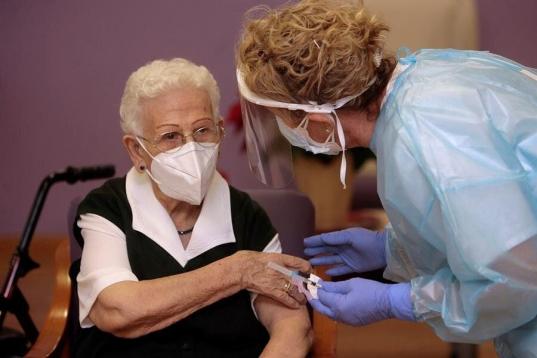
Araceli y Mónica, primeras personas en ser vacunadas contra el coronavirus en España.


Araceli, de 96 años, primera persona en ser vacunada de coronavirus en España.

Araceli, de 96 años, primera persona en ser vacunada de coronavirus en España.

Araceli, de 96 años, primera persona en ser vacunada de coronavirus en España.

Araceli, de 96 años, primera persona en ser vacunada de coronavirus en España.















